
|
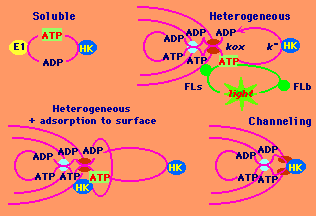
Four cases of cyclic coupled systems.
The systems include enzyme or mitochondria as bulk or surface ATP sources, and an ATP sink (e.g., hexokinase) present in solution, as well as a probe for ATP (firefly luciferase, FL) in solution or localized. It is expected that in heterogeneous coupled systems with high catalytic activity of one or both components, the physical steps (e.g., diffusion of nucleotides) may limit the total flux in the cycle. The steady state concentrations of ATP in solution and at the surface of mitochondria in experimental data have been mathematically modeled in each system. |

|
Mitochondrial compartments potentially subject to ATP monitoring by differently localized firefly luciferase probes. The system described include mitochondria as an ATP source, and an ATP sink (e.g., hexokinase) present in solution, as well as luciferase probes. The catalytic systems are coupled through diffusion of ADP/ATP. It is expected that concentration gradients of nucleotides occur in the vicinity of the mitochondrial surface. Differences in light emission by the different probes reflect the different local ATP concentrations sensed by each probe. Contact sites between mitochondrial membranes are believed to harbor a multienzyme complex including matrix ATP synthase, translocase, [creatine kinase], porin and hexokinase at the cytosolic surface, in which nucleotides are channelled in and out the mitochondrion. |
|
I'm a clickable imagemap...
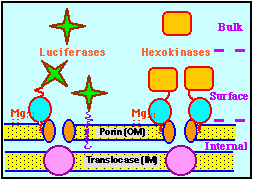
Localized hexokinase and luciferase species. Hexokinase (HK) exists naturally as a soluble (yeast) or a mitochondria-bindable (hepatoma, mammalian brain) form. The binding to porin at the outer membrane (OM) occurs through hydrophobic interaction with an N-terminal domain of HK. The complex may be stabilized by Mg-bridges and reversible oligomerization of HK in its bound state. Luciferase is soluble, but can be engineered so its chimeric products are anchored to the OM (lateraly delocalized) or porin (localized) by fusing the C-terminal luciferase moiety to N-terminal domains of OM-proteins or that of brain-HK, respectively. |
Molecular recognition - more info on the algorithm GRAMM and Ilya Vakser

|
Digitized trypsin-trypsin inhibitor complex.
The molecules have been digitized so that in the probed molecule, the core (black) is surrounded by two surface layers (blue) in the full representation (left), or a single layer adjacent to hydrophobic patches only (right). In the probing molecule (orange) all elements (left) - or only these representing hydrophobic atoms (right) - of the core are presented. A fitting score is calculated as the algebric sum of all weighted overlaps (positive) and penetrations (negative). |
|
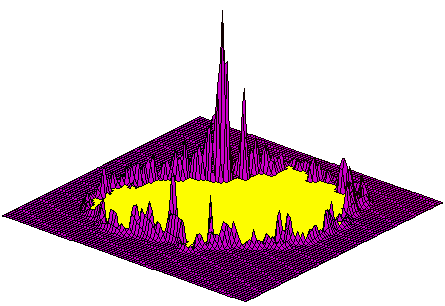
Correlation function.
Geometric surface complementarity calculated as the correlation function (using Fourier transformation). The plot represents a two dimensional section of the correlation (geometric maching score) as a function of the relative translation of the probing digitized molecule towards the probed molecule, at a given relative orientation (3D rotation). The central yellow "crater" represents negative values obtained by overlap between atoms in the respective cores of the molecules (penetration). The strong and distinct peak represents the best fit position, while the smaller spurious peaks around the crater represent random, non-specific contacts. |
Structure and evolution of mitochondrial porin (VDAC)
|
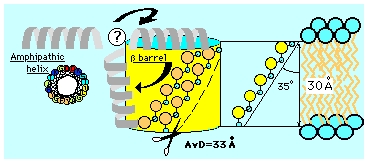
Schema for the mitochondrial pore.
The porin molecule has been modeled as a transmembrane beta-barrel by analogy to bacterial porins of known 3D structure, and based on molecular tomography. It creates a wide hydrophilic pore in the lipidic outer membrane, mostly beta strands with an amphipathic alpha helix at the N-terminus whose location is still uncertain (possibly dynamic allowing for pore size regulation). In order to visualize primary and secondary structure details, the cylindrical shape has been unfolded and projected on a plane as shown in the next figure. |
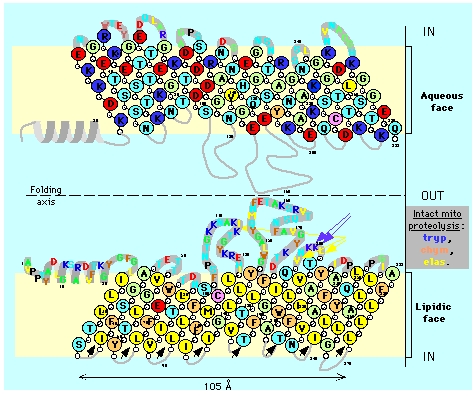
The aqueous and lipidic faces of porin modeled as a 16 TM beta strands barrel.
Such topology implies a special arrangement of amino acid residues in the sequence of transmembrane segments (beta strands or alpha helices) with polar residues facing the interior of the barrel and hydrophobic residues facing the external membrane phase. Two views of the planar projection of the porin molecule are presented, separated with an axis of symmetry (as a folding line). The up and down strands are connected by hydrophilic loops (gray) facing the cytosol (middle) or the inter-membrane space (top/bottom). Note the predominance of polar, acidic, and basic residues on the aqueous face and loops, while on the lipidic face hydrophobic, and aromatic residues are prevalent.

Sequence alignment of several mammalian hexokinase-binding porins.
The predicted model presented above is based on common features shared by porins. These can be revealed in part by examining their primary sequence (most often obtained from translation of the cDNA sequence). The alignment above, generated using the Clustal program (and manually adjusted to account for other experimental information) emphasizes a relative lack of sequence homology, while indicating strong potential similarity for secondary structure (amphipathic strands). Most the variability, including gaps (-), occurs in loops between transmembrane beta strands (red or yellow strips in two concurrent models). Key: HVDACx, human isoforms; MVDACx, murine; DMVDAC, D. melanogaster; WVDACx, wheat; YVDACx; S. cerevisiae.
Green algae: Physiology, Cellular and Molecular Biology
|
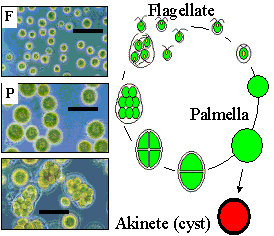
Life cycle of Haematococcus pluvialis.
Vegetative cell cycle of H. pluvialis. Left panel (bar = 50 mm): wild type small flagellated (F) cells soon loose their flagella, round up into palmelloid (P) cells of increasing size and divide within the mother cell theca (bottom). Right panel: cell cycle: under favorable conditions, round palmelloid green cells grow in size exponentially, and divide to yield flagellated, motile daughter cells (up to 16/mother cell), which soon loose their flagella and turn back to thick wall-bonded palmelloids, the most prominent form of the alga in non-synchronous cultures. When the conditions become unfavorable, the cells stop dividing, turn red (astaxanthin), increase in size and thicken further their cell wall (encystment) to become resilient cysts. Watch a presentation: H. pluvialis basic and stress physiology Watch another one: Carbon flux in green algae for biodiesel production |
|
|
|
|