In this section, you will learn to study models that contain more than one protein chain. For this section, obtain the file 3HHB from the PDB. Name it 3HHB.pdb.
Start SPdbV and open 3HHB.pdb, a model of deoxyhemoglobin.
Color: Chain
Many proteins are oligomeric -- composed of more than protein
chain, or subunit. SPdbV colors each subunit a different color. This
command is a quick way to find out how many chains are in a newly
opened PDB file.
Click on the Control Panel to activate it. Notice that letters A in the first column, the chain column. Click on any A, then press return. Clicking in the chain column selects the entire A chain. Scroll down to the end of the list of A-chain residues and click on any B, then press return. Chain B appears and chain A disappears. The chain column provides for quick and easy selection and deselection of entire chains. If the protein has only one chain, this column is empty. Clicking in an empty chain column selects the whole model.
Chains A and B in this model are the alpha and beta subunits of hemoglobin. You may be aware that hemoglobin consists of four subunits, two identical alpha and two identical beta. Some PDB files for oligomeric proteins contain only the unique subunits -- in this case, one each of alpha and beta. Later, you will build the additional subunits. But first, you will examine the interface between the subunits.
Color the chain A cyan (between blue and green on the color wheel) and the B chain magenta (between red and blue).
Select: Groups Close to another Chain...
Use this dialog to select groups that are within 5
angstroms of another chain, and click OK. This amounts to
selecting the residues within 5 angstroms of the subunit interface.
Press return to eliminate other residues.
Study the interactions in the subunit interface as follows. Color the selected sidechains (not backbones) by type, and then compute H-bonds. Zoom in for a close examination. The backbone colors allow you to distinguish chains A and B, while sidechain colors suggest the types of interactions. Red side chains (negative) near blue (positive) suggests ionic interactions. Gray near gray implies hydrophobic interactions. Green dotted lines indicate H-bonds. Do you see any interchain H-bonds? Measure any potential ionic or hydrophobic interactions. Slabbing and showing surfaces of selected groups may help you to see more about how the subunits interact.
When you are finished, redisplay the full model. Reset the Control Panel color menu to color backbone + sidechain, and color the model CPK. Turn off the display of H-bonds.
If you want to examine A-A or B-B interactions, you need a model of the entire tetramer. If the proper information is provided in the PDB file, SPdbV can help you build the full model. In the remainder of this section, your will build the full tetramer from two copies of the file 3HHB.pdb.
File: 3HHB.pdb (look for this file name
near the bottom of the File menu)
Color: Layer
This command, which uses SPdbV's list of recently used files,
loads a second copy of 3HHB.pdb. The second command colors the two
models different colors. SPdbV automatically gives the second copy
the same position and orientation as the first copy, so the new model
is invisible for the moment.
These two models are called layers. At the top of the Control Panel, notice the name 3HHB. The residues listed are those of the first 3HHB file that you loaded, which is currently the active layer. The name 3HHB is on a menu. Pull it down to see that 3HHB is listed twice. Select the second 3HHB on the list. Now you will rename it so you can more easily tell the two layers apart.
Edit: Rename Current Layer...
Name the new layer 3HHBA'B' and click OK.
Click the checked button labeled visible just below the file name on the Control Panel. The model 3HHBA'B' (blue) disappears, and you can see the model 3HHB (yellow). The visible button allows you to turn model display on and off without having to close the file. Click the button again to redisplay 3HHBA'B'. Move the two models around using the rotate and translate functions on the graphics window. You may see the display flicker between the colors of the two models.
Now click on the can move button, and try again to move the models. The yellow model, 3HHB, moves, but 3HHBA'B' does not. Use translation to move 3HHB away from 3HHBA'B'. Click the can move button again to restore movement of 3HHBA'B'. The can move button allows you to turn individual model movement on and off.
Edit: Reset Orientation (current layer
only)
Make 3HHB the active layer.
Edit: Reset Orientation (current layer only)
Now the layers are again superimposed. Make 3HHBA'B' the active
layer, and select chain A.
Edit: Rename Current Layer...
Tab into the box labeled Rename Chain of Selected Groups,
type C, and click OK.
In the 3HHBA'B' layer, select chain B.
Edit: Rename Current Layer...
Tab into the box labeled Rename Chain of Selected Groups,
type D, and click OK. You have renamed the A and B chains of
the new 3HHBA'B' layer so that when you merge both files into one,
the four chains will be named A and B (the first alpha/beta dimer), C
and D (the second dimer). Now you will rotate the CD pair into the
orientation of the second alpha/beta dimer.
Select:All
With all groups in the new layer (3HHBA'B') selected, click the
file icon on the graphics window (beside the little earth icon) to
display the PDB file. Scroll down to the beginning of the ATOM lines.
The last three lines, labeled MTRIX, before the first ATOM line give
the information that SPdbV needs to transform the coordinates of your
CD dimer into the coordinates of the second, or A'B' pair. All you
need to do is click anywhere in these three lines, and click
OK on the dialog box. Then close the file window, and you will
find the current layer in its new orientation, with the original
layer in the old orientation. Together, they make a full tetramer.
Now you will combine them into a single layer, and then save the
tetramer in a new file.
<shift>Select: All (selects all in
both layers -- recall that shift applies
Select commands to all layers)
Edit: Create Merged Layer From Selection
This operation will take a few seconds. Make the layer 3HHBA'B'
invisible. Then make the layer 3HHB invisible. Notice that you now
have a third layer, named "_merge_" . Make "_merge_" the active the
layer.
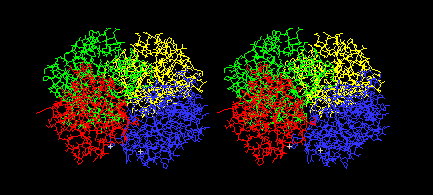
Color: Chain
You should see the four chains of the tetramer in different
colors, like this:
File:Save As...
Save the file as 3HHBTetramer.pdb.
Close all files and then open your newly saved file. Rotate and study the way the four oligomers fit together.
Select: Groups Close to Another Chain
Use the dialog that appears to select groups that are within 5
angstroms of another chain, and click OK.
<return>
Now you can study all of the subunit interfaces of the
tetramer.
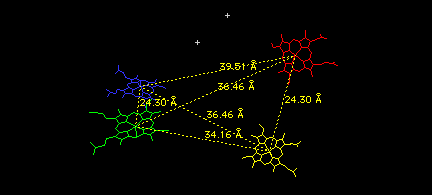
Finally, look at the relative positions of the four hemes (and two phosphate ions) in the tetramer as follows:
Select: Group Kind: HETATM
<return>
You should see only the four hemes and two phosphate ions. Use the measurement icon on the graphics window to measure all the distances between iron atoms in the various hemes. When you have measured them all, the six yellow lines will form an elongated tetrahedron, with its shortest sides connecting the two alpha/beta dimers you have combined to build this tetramer.
Close all files.