In this section, you will alter the model by changing an isoleucine residue to glutamine. Then you will investigate whether the new residue might form an additional H-bond to tri-NAG. This is the kind of study a researcher might carry out while trying to redesign a protein to make it bind its substrate more tightly.
You will find this operation much easier in stereo. If you have not learned to view in stereo, this is a good time to work on it. Click here for help.
Display and center the complete model 1HEW in CPK colors, and display H-bonds. (If you have stopped and restarted SPdbV since you last displayed H-bonds, you may have to compute them again.) Then restrict the display to tri-NAG and groups within 7 angstroms of tri-NAG. Display only H-bonds to tri-NAG.
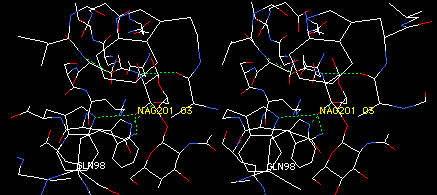
On the Control Panel, click in the label column next to ILE98. Find ILE98 on the display, and note that its side chain reaches in the general direction of tri-NAG, making it a candidate for engineering additional hydrogen bonds. Leave ILE98 in the clear so you can click on one of its atoms in the next operation.
Click the MUTATE button (second from right in button row), and then click any atom of ILE98. From the pop-up menu that appears, click on GLN to replace the isoleucine with glutamine. (Glutamine is a good choice because it will provide a long side chain with the capacity to either donate or accept an H-bond). Now examine the various staggered conformations (rotamers) of this side chain by repeatedly pressing the * key on the numerical keypad, or by clicking on the small arrows under the mutate tool icon. With each press or click, SPdbV shows a different rotamer, and also shows you clashes (pink dashed lines) and potential H-bonds (green) between the side chain and other parts of the molecule. Try to find a rotamer that makes an H-bond to tri-NAG, and that does not clash into other residues. You should find a rotamer with a potential H-bond to atom O3 of NAG201. Leave the side chain in this position. Your display should look something like this:
Notice that the MUTATE button has remained darkened during these operations, because the changes you are making are still pending -- SPdbV has not saved your changes yet. Click the button again and click Accept to keep the new GLN residue in the conformation you selected.
Now you will alter this rotamer manually to see if the new side chain can reach any other atoms. Click the TORSION button and click on any atom of the new GLN98 residue. Now you are ready to change the conformation of this side chain. Note that the TORSION button remains dark, indicating that the changes you are making are pending, and you will get a chance to accept or reject them.
Hold down the 1 key (the number one) on the keyboard while dragging the mouse to the left or right. This action rotates about the first bond (CA-CB) of the side chain. To rotate about the second bond (CB-CG), hold down 2 and drag the mouse, or 3 for CG-CD. In this manner, you can adjust the conformation of the three rotatable bonds of the GLN side chain. As you rotate the atoms around a bond, lines indicating clashes or potential interactions appear and disappear. See if you can find a conformation in which a side chain atom of GLN can form a hydrogen bond with NAG 202 O6, without clashing with other atoms. With some fiddling, you may even be able to make the O or N of GLN bridge NAG 202 O6 and NAG201 O3.
NOTE: SPdbV shows only realistic (staggered) rotamers during the MUTATE function. But during TORSION functions, SPdbV does not check your modified side-chain conformations to be sure they are realistic. Mutating and changing torsion angles manually can produce models that are chemically impossible. In practice, you would follow up such alterations to models by minimizing the energy of the model to eliminate unreasonable features. This requires an external program such as AMBER, CHARMm, or GROMOS. If you have access to these programs, you can learn how to submit jobs to them from SPdbV in the advanced tutorial on energy minimization.
To accept the changes you've made, click the darkened TORSION button again, and click Accept. Now SPdbV has recorded your changes in the coordinate file. Now save your hard-won mutant form of lysozyme, as follows:
File: Save As ...
Navigate to save this file on your own floppy disk.
Then name the file 1HEWI98Q.pdb. Click OK to complete the
save. Do you know what this file name means?
Now notice that the Control Panel and graphics windows still display the original name of the file. But the file itself, 1HEW.pdb, is safe and sound in its original location. SPdbV protects your original files by making it hard for you to save changes in them accidentally. The program contains no simple Save command that can overwrite an existing file without giving you a warning.
Take a look at the various options for saving, under the File menu. SPdbV allows you to save your entire file, or just selected residues. In either case, when you reopen these files, you will find most settings unchanged, and your last view restored. So saving a file provides a way to get back to your previous task without repeating any operations. In addition, you can save a text file of the amino-acid sequence of your model, in a format you can use for searching for proteins of similar sequence.
For more information on these functions, click on SPdbV User Guide in the Contents frame (at left), and read the Tools and Mutation sections. Also see Tips and Tricks, and read the Preferences section to see how you can customize SPdbV to suit you better. Learn more about preferences by looking at some of the dialog windows that come up when you pick commands under the Preferences menu.
In normal operations, SPdbV saves your current settings as the default preferences for future sessions. But you can also create and save several sets of preferences that you find handy for certain operations. You can save your current preferences with the Preferences: Save As ... command. Give your preferences a name like YourName.prf. During SPdbV operation, if you open your preferences file with Preferences: Open, SPdbV will adopt your settings for the rest of your session.
Take time to PLAY with the tools introduced in this section.